在药物研发的全生命周期中,安全性评估不仅是受试者保护的基石,更是监管合规的红线。自2019年我国实施《ICH E2F:研发期间安全性更新报告指导原则》以来,DSUR已成为申办者每年必须面对的一场“年度大考” 。然而,在实际操作中,如何将军际标准与中国区域监管要求(如2020年发布的《管理规范》)深度融合,依然是许多RA、PV及临床申办方的痛点 。
2026年2月,国家药监局药审中心(CDE)发布了最新的《研发期间安全性更新报告的问答文件》,针对行业关注的热点与难点进行了系统解答 。为了帮助从业者更直观地理解这些变化,我们将这份厚达十几页的官方文档提炼为这份“实操蓝图”,旨在填平理论与执行之间的鸿沟 。
本期内容核心亮点:
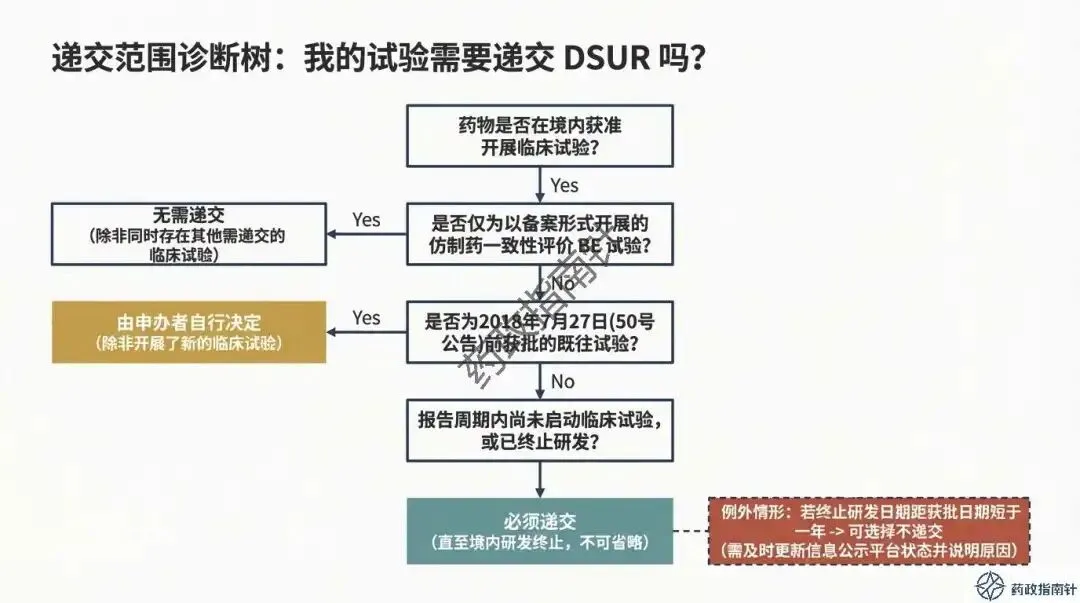
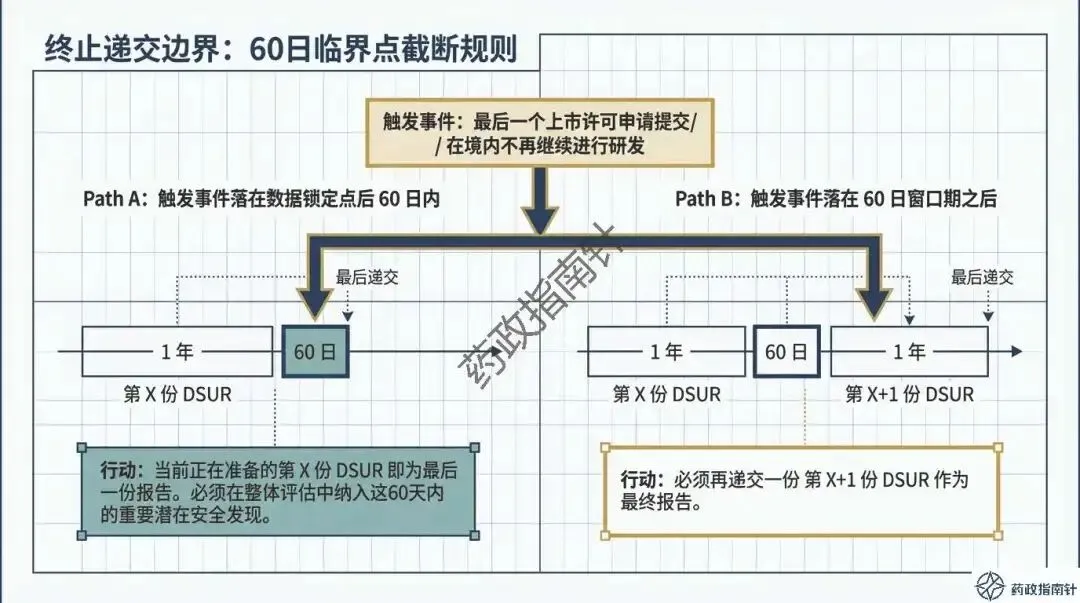
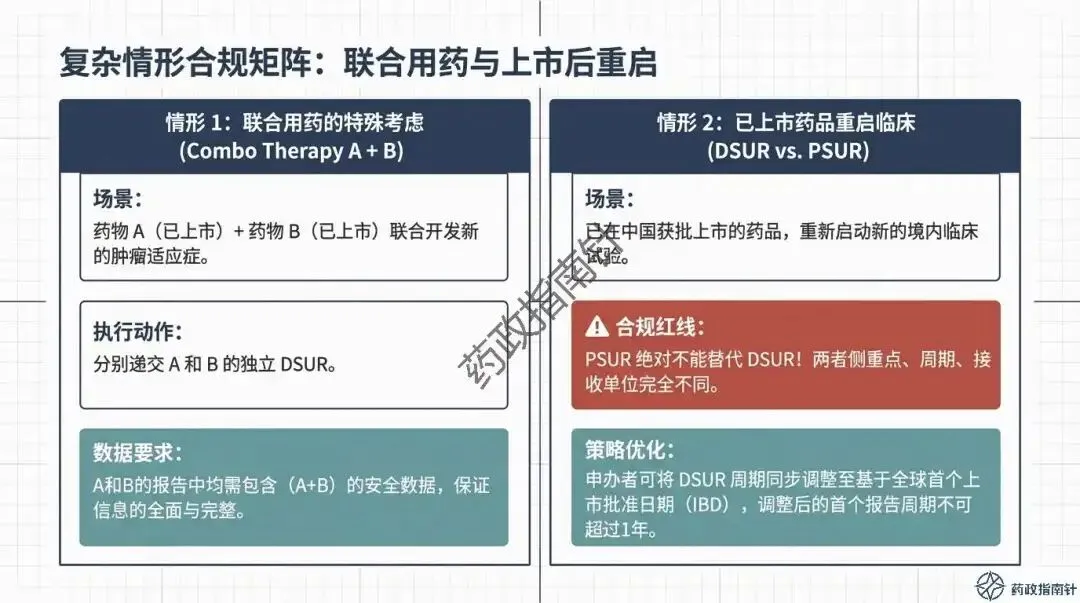
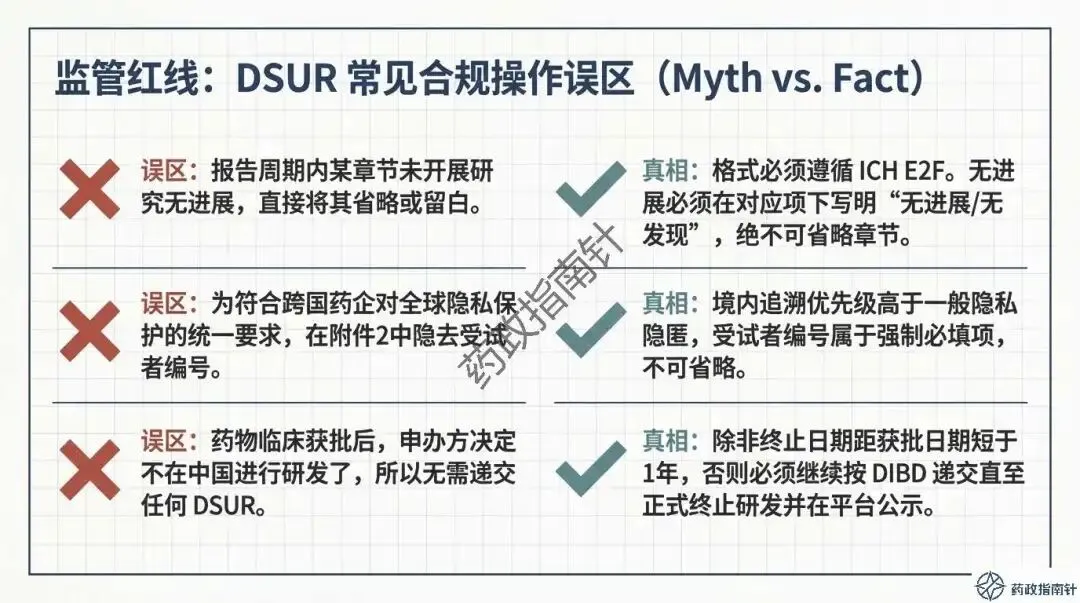
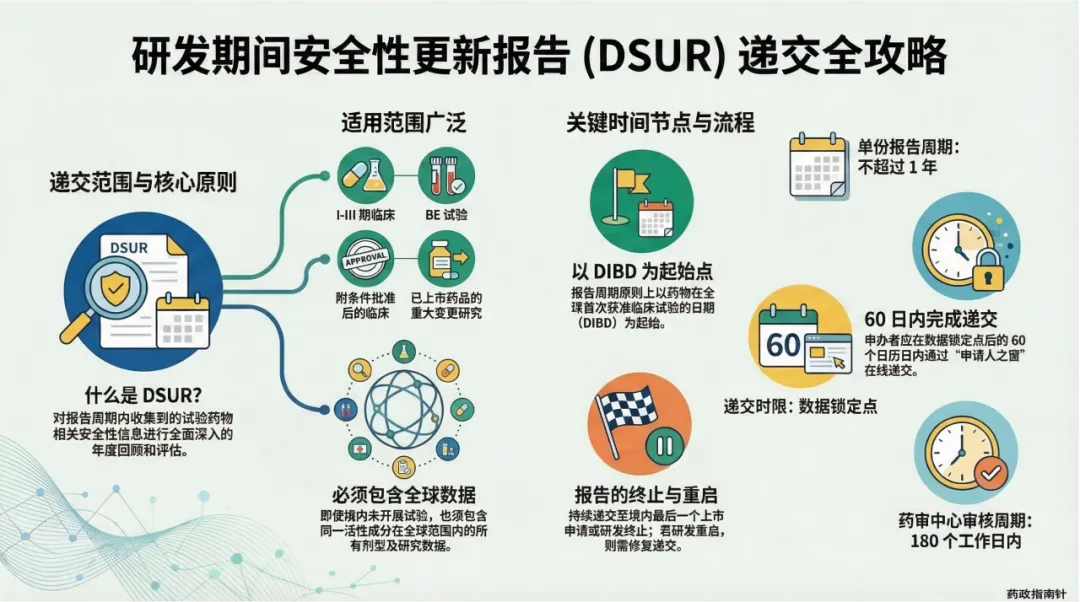
递交边界的精准划定:明确了境内获准临床试验的递交范围,特别是针对仿制药一致性评价BE试验、2018年前获批的老项目以及研发终止等特殊情形的递交判定 。
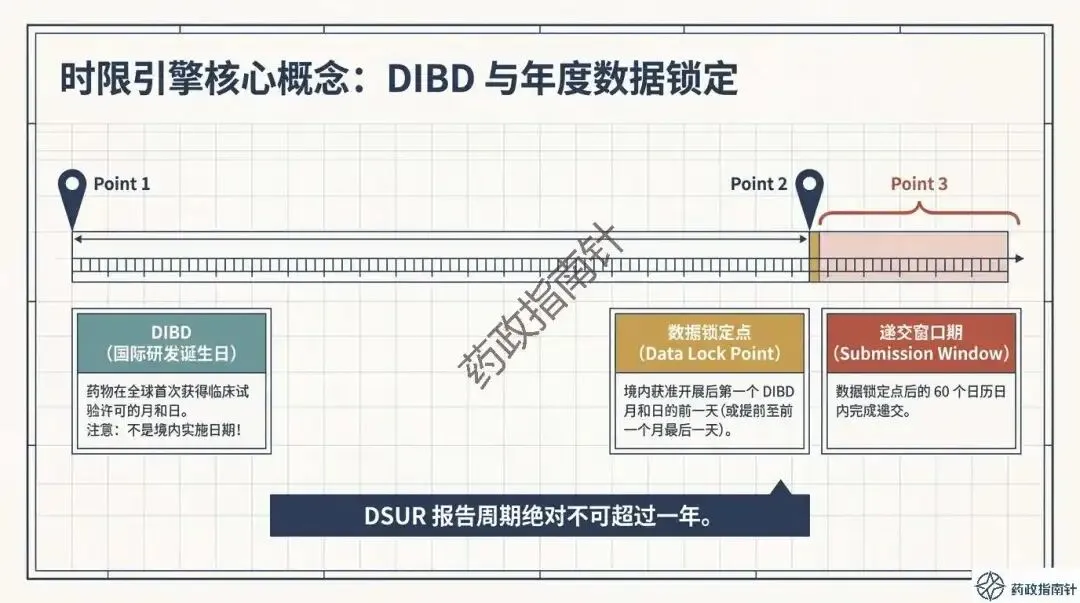
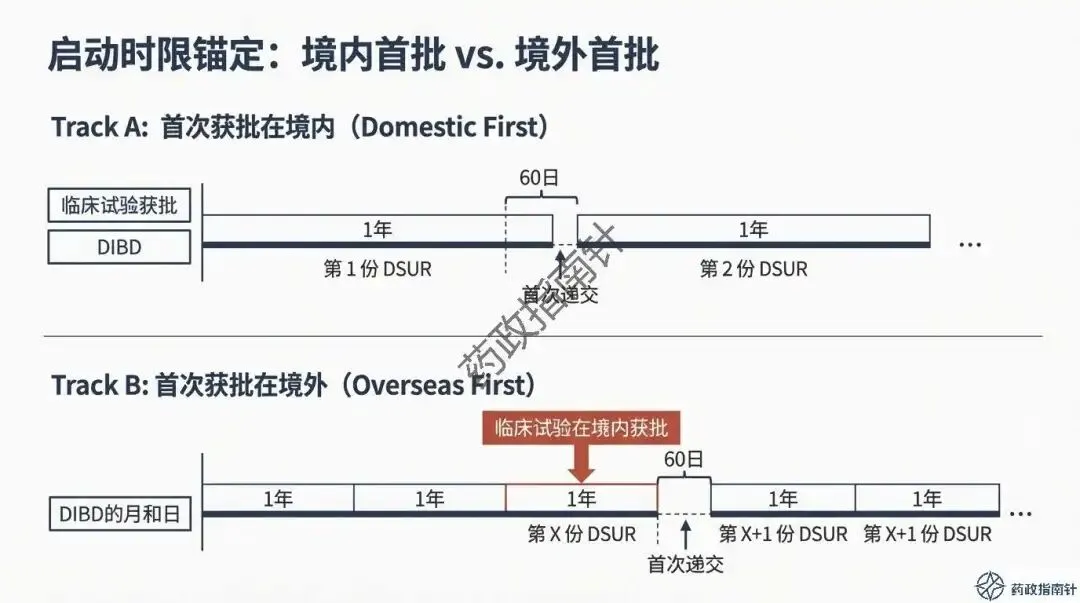
时限引擎的深度解析:详细解读DSUR的报告起始时间(国际研发诞生日期,IBD)与截止时间,确保申办者不再错过关键的时间节点 。
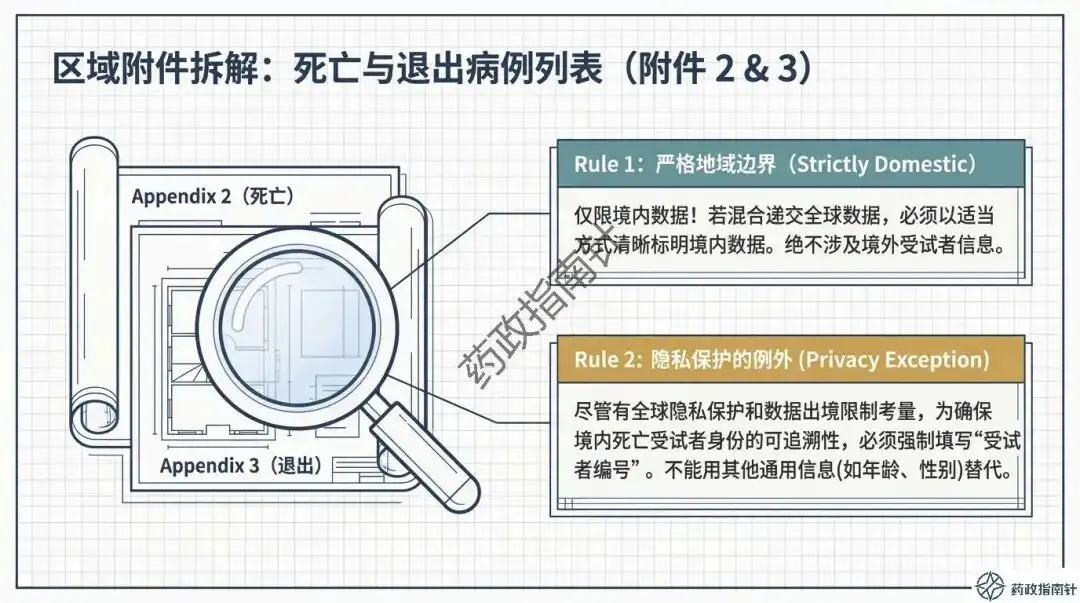
区域附件的合规细节:重点剖析了“中国特色”附件的要求,包括境内死亡病例列表的填写规范,以及在隐私保护前提下如何确保受试者编号的可追溯性。
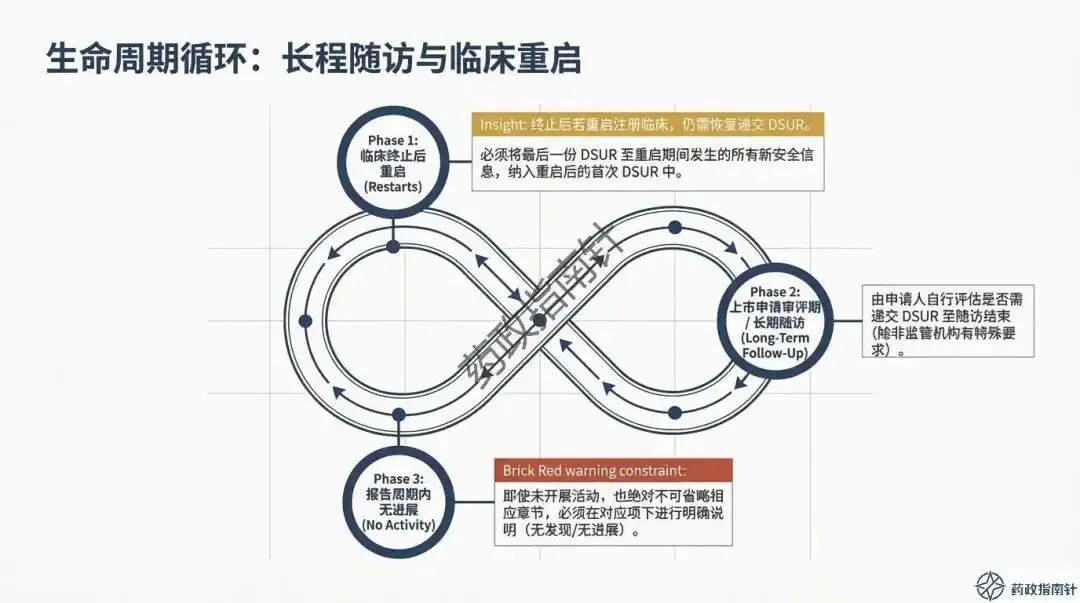
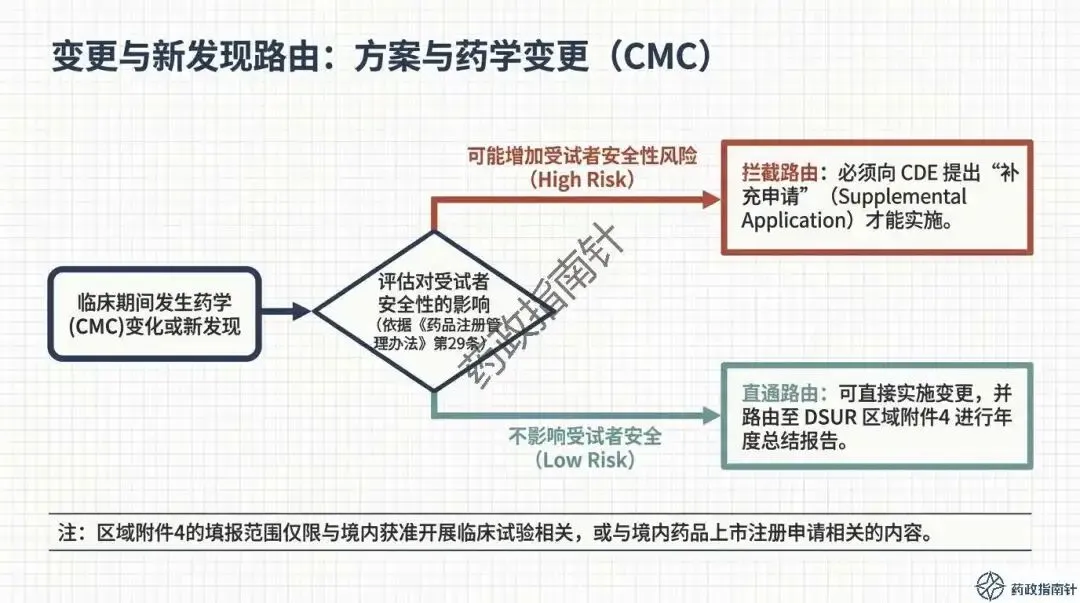
变更路径的合规路由:当临床期间发生药学变更(CMC)或方案调整时,究竟该通过DSUR报告还是递交补充申请?我们为您梳理了清晰的判断逻辑 。
DSUR的核心目的,是对报告周期内收集到的相关安全性信息进行全面深入的年度回顾 。它不仅是一份合规文件,更是一份动态的“获益-风险评估报告”。接下来的PPT内容,将带您用PPT读懂2026年DSUR的最新监管脉络。
DSUR的工作绝非简单的资料堆砌,而是一场跨部门协作的科学评价过程。
在后续的实操中,我们建议关注以下三大合规边界:
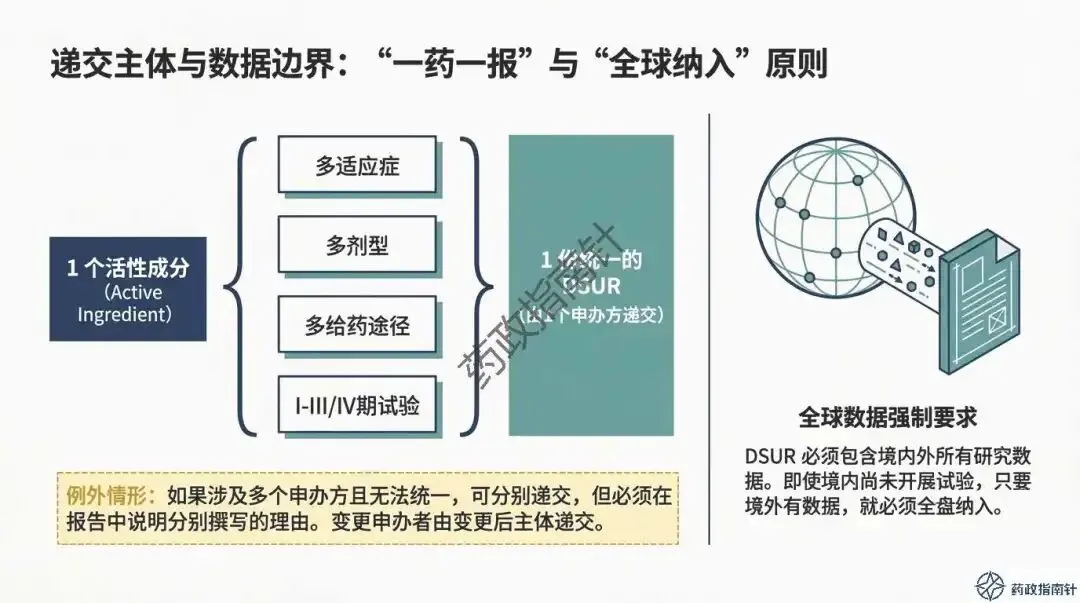
首先是数据的地域准确性。在区域附件的准备中,CDE有着明确的“严格地域边界”要求。例如,附件2应仅限中国境内死亡受试者信息,而非全球数据的简单罗列 。若混合递交,必须清晰标明境内数据,这是确保监管审核效率的关键 。
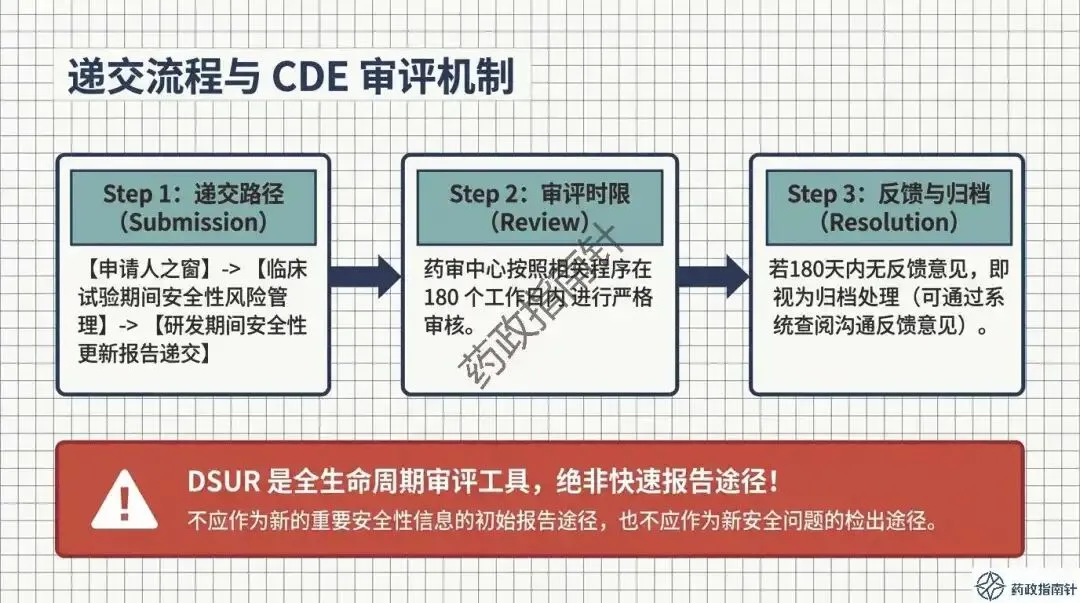
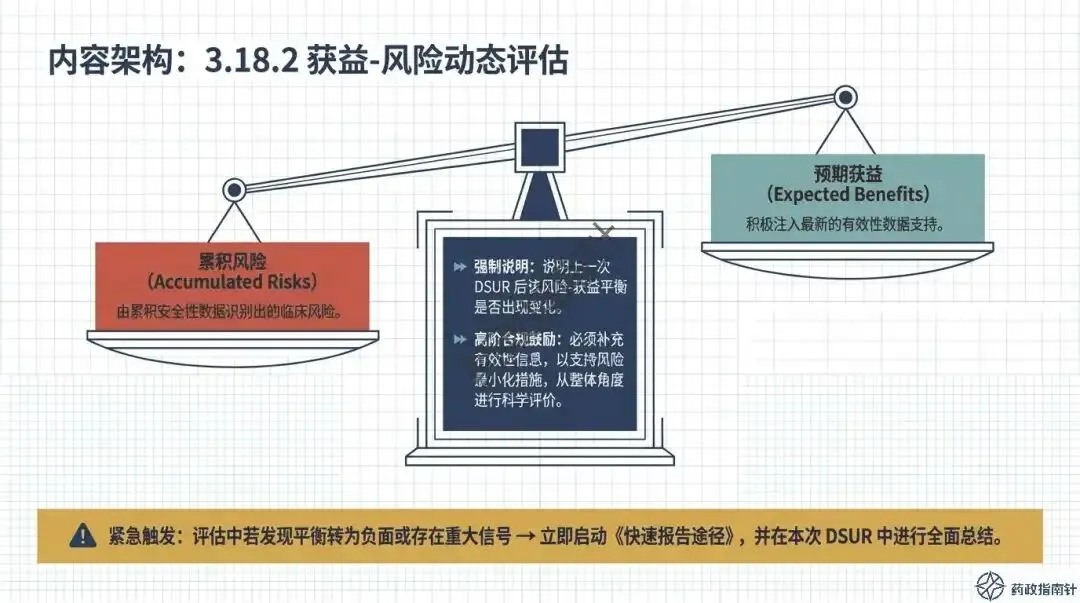
其次是动态风险评估的深度。根据最新的合规鼓励方向,申办者不应仅列出累积风险,更应积极注入最新的有效性数据支持,进行“获益-风险动态评估” 。如果评估中发现风险-获益平衡转为负面,或者出现了重大安全性信号,绝不能等待DSUR的年度报告,而必须立即启动《快速报告途径》 。
最后是变更管理的审慎性。对于可能增加受试者安全性风险的药学变更,应严格按照《药品注册管理办法》提出补充申请,而非仅在DSUR中予以告知 。DSUR更像是一个“汇总窗”,记录那些评估后认为不影响受试者安全、可直接实施的变更 。