资料解读:《新版GMP质量管理体系培训》
详细资料请看本解读文章的最后内容。
本次培训围绕新版GMP(药品生产质量管理规范)质量管理体系展开,内容涵盖了新版GMP的核心特点、质量风险管理、无菌附录要求以及认证检查等多个关键模块,旨在系统提升药品生产企业的质量管理水平与合规实践能力。
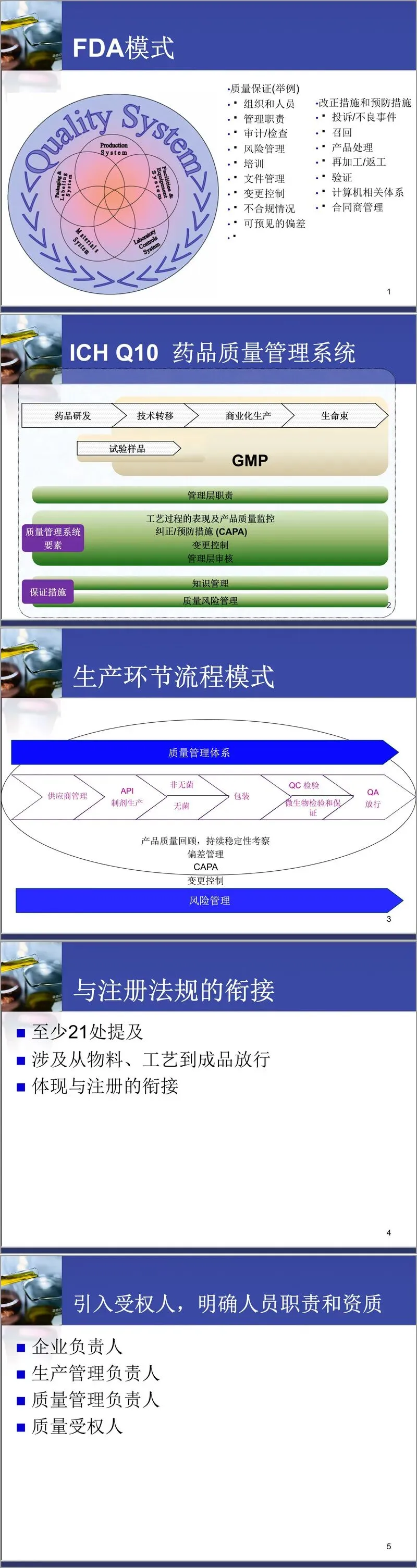
培训首日由肖志坚主讲,重点介绍了新版GMP的总体特点。新版GMP强调质量管理体系的系统构建,注重与药品注册法规的衔接,明确引入了“质量受权人”制度,进一步强化了关键人员的资质与职责要求。同时,风险管理理念被正式纳入体系,要求企业在前瞻或回顾的基础上,对质量风险进行评估、控制、沟通与审核。此外,新版GMP还对委托加工与委托检验提出了明确管理要求,以适应行业协作发展趋势。
在质量管理体系部分,文件明确指出,质量管理体系是企业为保障产品、过程或服务质量,将组织机构、职责权限、工作方法、技术资源、信息等要素整合而成的有机整体。培训中借鉴了FDA模式与ICH Q10标准,列举了质量保证的关键环节,如组织与人员、管理职责、审计检查、文件管理、变更控制、偏差处理、CAPA(纠正与预防措施)、投诉召回等,并强调这些环节需在知识管理与质量风险管理的框架下协同运作。
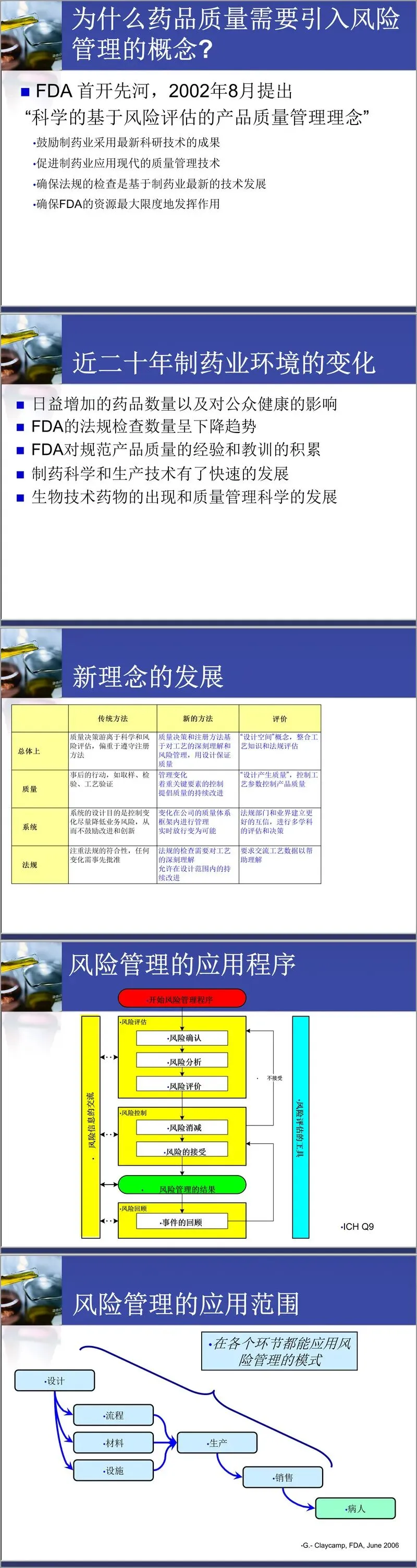
质量风险管理是本次培训的核心议题之一。培训从风险的定义入手,指出风险是坏事发生的概率与后果严重程度的综合。ICH Q9为风险管理提供了方法论基础,强调风险管理应贯穿药品全生命周期——从研发、临床、生产到销售。新版GMP明确要求企业运用风险管理工具,如流程图、检查表、失效模式与影响分析(FMEA)、危害分析与关键控制点(HACCP)等,在工艺验证、环境监测、偏差调查、稳定性研究等方面实施风险控制。培训特别指出,风险管理的重要性不仅在于识别风险,更在于通过团队协作、透明决策和预防措施,实现资源的合理分配与质量体系的持续改进。
无菌药品附录是本次培训的另一重点。新版无菌附录在硬件与软件方面提出更高要求,洁净级别与国际标准(如ISO 14644)接轨,明确了A级、B级区的动态与静态标准,增加了吹灌封及隔离操作等技术章节,并强化了全过程环境监控。培训详细阐述了洁净区划分、粒子监测、自净时间、培养基模拟灌装试验、轧盖环境、人员控制等方面的具体要求。其中,培养基灌装试验作为无菌工艺验证的核心手段,其接受标准、试验设计、最差条件挑战及失败调查等内容均被深入探讨。此外,培训还强调了过滤器验证、容器密封性验证、共线生产风险评估与清洁验证的重要性,确保无菌药品在生产全过程中不受微生物、热原与微粒污染。
培训后续部分由华蕾与季铁军分别讲授,内容涉及GMP认证检查、自检、变更控制、微生物室管理、固体制剂、偏差处理、年度质量回顾等实务环节。认证检查强调企业应建立完善的自检体系,确保持续符合法规要求;变更控制需基于风险评估,确保任何变更不影响产品质量;偏差管理要求企业建立分级调查机制,从根本上解决问题并采取预防措施;年度质量回顾则是对全年生产质量数据的系统分析,以评价工艺稳定性与质量趋势。
总体而言,本次培训体系完整、内容深入,既突出了新版GMP的理论创新,又提供了扎实的实践指导,尤其强调风险管理的融入与无菌控制的强化,为中国制药企业提升质量管理能力、接轨国际标准奠定了重要基础。
接下来请您阅读下面的详细资料吧。