辅助供氧是现代临床诊疗的核心干预手段,广泛应用于急诊、围手术期及危重症患者低氧血症的纠正。尽管氧疗能够挽救生命,但越来越多的研究证据表明,过度氧暴露会引发显著的病理生理紊乱,尤以心血管系统和呼吸系统受累最为明显。高氧血症(动脉血氧分压>100 mmHg)会促进活性氧生成,进而诱发氧化应激、线粒体功能障碍及促纤维化通路激活。当联合机械通气使用时,胸腔内压力改变、静脉回流减少、肺血管阻力升高等效应会进一步被放大,最终使心肌承受血流动力学负荷。机械作用与生化紊乱共同介导心脏结构、功能及电生理重构,引发传导异常与心律失常。
来自危重症及新冠感染人群的最新临床研究提示,精准滴定式氧疗策略至关重要,需在保证组织充分氧供的同时,最大程度减轻高氧损伤。本文综述高氧诱导氧化应激、心肺交互作用及心肌重构的相关机制,为优化临床氧疗方案提供全面的理论框架。
关键词:辅助供氧;高氧血症;肺生理;心肌重构;心肺交互作用;氧化应激;机械通气;活性氧
辅助氧疗是现代医学中应用最广泛的干预措施之一,也是低氧血症、呼吸衰竭及危重症患者的核心治疗手段。通过提升动脉血氧含量,辅助供氧可维持组织氧供、预防器官功能损伤。临床数据显示,约半数重症监护病房住院患者在治疗过程中均会接受氧疗干预。
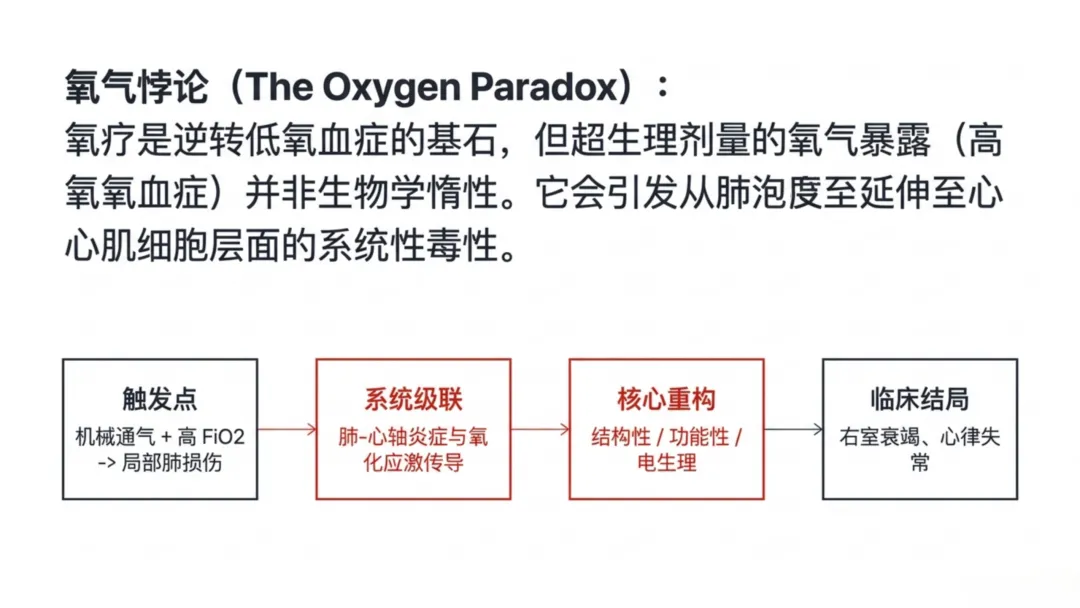
但氧气并非生理惰性物质,大量研究证实,过度氧暴露会破坏细胞稳态,诱发全身毒性损伤。氧毒性的核心机制为高氧血症,临床将动脉血氧分压超过100 mmHg定义为高氧血症诊断临界值,其临床危害与严重程度密切相关。轻度高氧血症(动脉血氧分压 100~200 mmHg)常出现于常规供氧过程中,健康人群通常可耐受;中度高氧血症(动脉血氧分压 200~300 mmHg)会诱发进行性氧化应激、早期内皮功能损伤及明显血流动力学改变;重度高氧血症(动脉血氧分压>300 mmHg)多由机械通气过程中高浓度吸氧所致,肺与心血管毒性风险最高,也是本文病理生理机制探讨的重点范围。
高氧血症本质是血液及组织氧水平超出生理需求,多由辅助供氧或机械通气时高浓度吸氧引发。高氧环境下,氧气发生不完全还原,生成超氧阴离子、过氧化氢、羟自由基等活性氧。过量活性氧会超出机体抗氧化防御能力,造成氧化损伤,诱发肺、血管及心肌损伤。
危重症患者体内,氧疗与心肺生理的交互作用尤为复杂。机械通气联合高浓度氧气可改变胸腔内压力、肺血管阻力及心室负荷状态,进而影响心输出量与心肌应激水平。除血流动力学改变外,高氧血症还会激活炎症信号通路、诱发线粒体功能障碍,参与心肌重构与电生理不稳定的发生。上述过程体现了心肺系统的紧密关联性,肺部损伤可通过炎症介质与氧化应激通路引发全身效应。
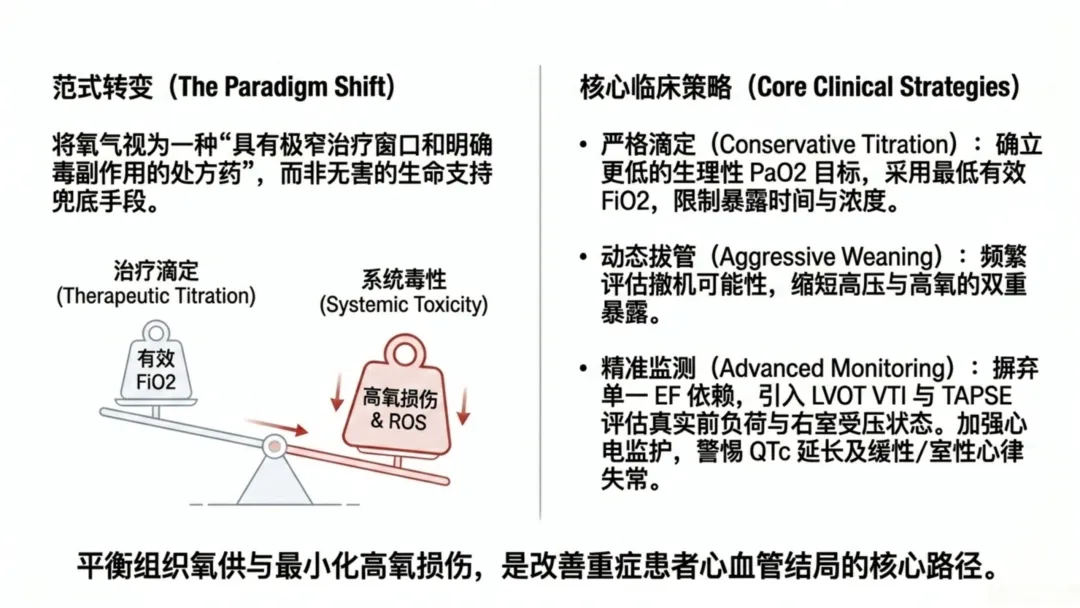
近年来临床与基础研究逐渐转向保守化、精准化供氧理念,在维持有效氧合的同时规避高氧损伤。深入阐明辅助供氧对心血管功能的生理影响,是优化危重症患者氧疗策略的关键。
本文系统阐述辅助供氧与高氧血症对心血管生理的影响,重点分析机械通气下的心肺交互作用、心肌结构重构机制、血流动力学功能改变,以及过度氧暴露相关的电生理紊乱。
本文为针对辅助供氧对心血管及心肺生理影响的叙述性综述。通过PubMed数据库,以高氧血症、辅助供氧、机械通气联合肺生理、姑息治疗、心血管生理为检索词开展全面文献检索。
纳入标准:研究直接关联心肺预后,优先选取随机对照试验及高影响力观察性研究;排除标准:技术报告及未经过同行评审的评述类文献,以保证综述学术严谨性。
辅助供氧是急诊与重症医学的基础治疗手段,核心用于逆转呼吸衰竭所致低氧血症。近半数重症监护病房患者需接受氧疗,临床诊疗的关键难点在于平衡氧疗获益与氧毒性风险。
辅助供氧可提供高于空气氧浓度的气体以改善气体交换,现行临床指南推荐采用滴定式供氧,根据胸廓起伏、呼吸音等床旁呼吸评估结果选择供氧方式。
临床供氧遵循由无创到有创的阶梯式方案,根据氧合需求逐级升级支持模式。无创供氧方式包括鼻导管吸氧与简易面罩吸氧:鼻导管吸氧流量1~6 L/min,吸入氧浓度可达24%~40%;简易面罩吸氧流量5~10 L/min,吸入氧浓度为40%~60%。随着氧流量与支持级别提升,通过无重复呼吸面罩、高流量供氧设备可将吸入氧浓度上调至接近100%。
无创通气为急诊科及院前急救一线方案,适用于慢性阻塞性肺疾病/哮喘急性加重伴高碳酸血症呼吸衰竭、肺水肿,以及移植术后、肿瘤等免疫低下人群低氧性呼吸衰竭,可在避免气管插管的前提下改善病情。
临床指南推荐急性低氧性呼吸衰竭患者优先选用高流量鼻导管吸氧,其可减少解剖死腔、提供加温湿化气体,同时提升患者舒适度与耐受性。高流量鼻导管吸氧也可用于无创通气间歇期维持氧合,但需严格评估病情,避免延误病情恶化患者的气管插管时机。
当高流量供氧无法满足氧合需求时,无创通气可降低呼吸肌做功、促进肺泡复张、维持气道开放,规避机械通气相关并发症。
有创通气需行气管插管或气管切开,机械通气可提供最高100%的吸入氧浓度,常联合呼气末正压为危重症患者提供气体支持。高浓度吸氧易诱发动脉及组织高氧血症。动物实验证实,机械通气联合100%纯氧会升高膈肌血管阻力,降低膈肌血流量与氧供,是高氧相关膈肌损伤的重要机制。
研究显示,50%浓度的辅助供氧可显著改善慢性充血性心力衰竭患者运动耐量、减轻劳力性呼吸困难,提示适度供氧可缓解通气与血流动力学限制,但仍存在心血管不良影响。心力衰竭患者常常规接受辅助供氧,而高浓度氧气会急性降低心输出量与每搏量,升高肺毛细血管楔压,对基础心功能储备较差的患者造成明显血流动力学负荷。此外,吸入浓度高于21%的氧气会通过迷走神经介导减慢心率,进而以心率依赖方式降低心脏指数。
高氧血症指机体组织与器官氧水平异常升高,多由高浓度吸氧或动脉血氧分压≥100 mmHg所致。高浓度氧气会通过过量生成活性氧产生毒性作用,高氧环境下肺、中枢神经系统、眼部损伤最为显著。氧毒性的发生时间与严重程度取决于氧暴露剂量和时长。氧悖论是高氧损伤的重要特征:缺血组织突然复氧会触发特殊损伤反应,造成的组织破坏远超缺血本身。
高氧血症分为常压高氧血症与高压高氧血症两类。
常压高氧血症:常压环境下给予高浓度氧气,通过面罩或呼吸机支持实现,广泛用于急性缺血性脑卒中治疗,可提升脑组织氧水平,增加缺血半暗带动脉血氧分压,保护半暗带组织,且不会显著增加氧化自由基损伤。
高压高氧血症:高压氧治疗过程中产生,患者在高于标准大气压的密闭舱内吸入近100%纯氧。高压氧可促进创面愈合,但氧气毒性进展更快,血液与组织溶解氧含量大幅升高,加速有害氧自由基生成,加重肺、脑等脏器应激损伤。肺部首发症状为气道刺激、咳嗽、黏液清除能力下降,持续暴露可进展为肺部炎症、肺水肿、胸痛及呼吸困难;同时可通过大脑与迷走神经通路诱发全身症状,体现直接氧损伤与神经系统调控的双重作用。
高氧血症可诱发多器官全身损伤。肾脏层面,过度供氧会改变肾血流量与氧稳态,参与急性肾损伤发生;心肺肾多器官交互作用会放大损伤效应,单一器官受累可继发其他器官功能障碍。同时,高氧血症是心肌重构的重要驱动因素。机械通气联合长期高氧暴露可损伤肺上皮细胞,动物实验显示小鼠持续高氧暴露72~96小时后死亡率升高,且体重下降10%~15%。本团队动物研究证实,高氧血症可延长QTc、JT间期,降低心输出量与射血分数,诱发心动过缓及心律失常。
高氧血症也存在一定临床获益,可通过提供高组织氧张力促进术后创面愈合,助力中性粒细胞介导的活性氧生成、吞噬作用及杀菌活性。临床需精准把控辅助供氧剂量,在满足治疗需求的同时规避全身氧化损伤风险。
机械通气是多种危重疾病的核心支持手段,核心目标是维持气体交换、保障全身氧合及二氧化碳清除,而吸入氧浓度的精准滴定是其中关键环节。
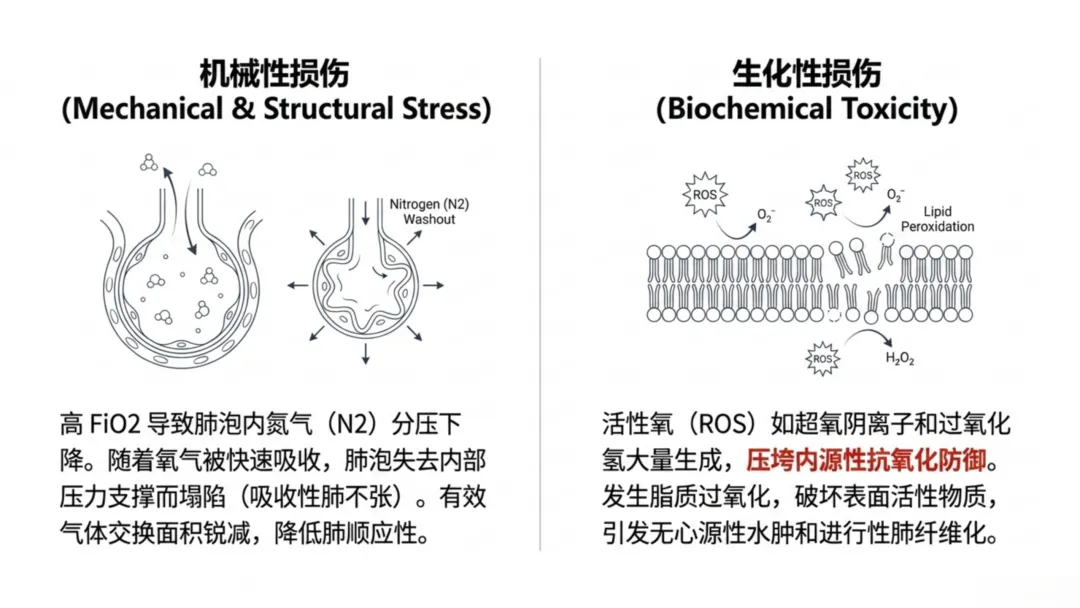
长期以来临床普遍采用宽松式供氧策略,规避细胞缺氧的即时风险,但现有证据证实,肺部对氧浓度高度敏感,供氧过量与供氧不足同样具有危害。高浓度氧气会改变肺泡力学稳定性,氮气作为肺泡正常结构支撑气体,被氧气冲刷排出后会引发吸收性肺不张,减少气体交换面积,进而被迫提升通气压力与氧浓度,形成恶性循环。
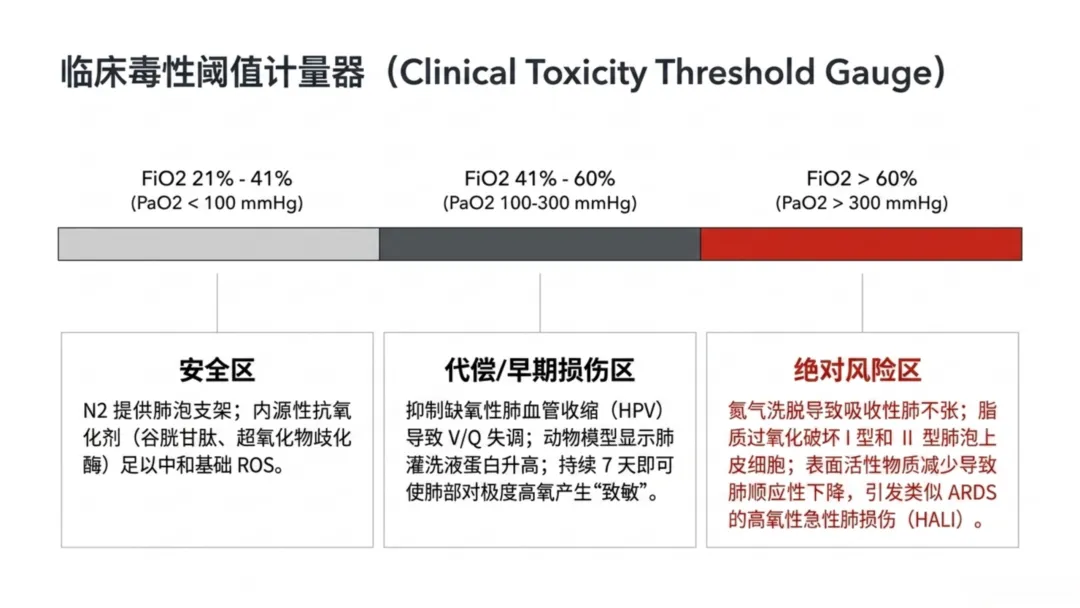
健康人体呼吸空气氧浓度为21%,机械通气患者将吸入氧浓度维持在21%~41%为安全区间,可维持血氧饱和度92%~96%的理想水平,此范围氧化应激风险极低,肺组织内谷胱甘肽、超氧化物歧化酶等抗氧化物质可有效中和活性氧。氮气在此区间可充当"支架",维持呼气末肺泡开放状态。
当吸入氧浓度升至41%~60%时,肺泡内氮气分压下降,氧气被肺毛细血管血红蛋白快速摄取,若氧气摄取速率超过肺泡通气速率,会导致肺泡容积缩小、肺泡塌陷。该浓度区间会抑制缺氧性肺血管收缩反射,正常生理下该反射可使血流从通气不良肺组织分流至通气良好区域;而中度高氧会使通气不良区域血管扩张,增加静脉掺杂率,降低气体交换效率。
基础研究显示,轻中度高氧(吸入氧浓度21%~60%)会升高肺泡灌洗液蛋白浓度,提示早期肺损伤;持续7天50%~60%浓度供氧,会使肺组织对后续高浓度氧毒性更易感。在感染及非感染炎症模型中,轻中度高氧会升高肺组织重量,是肺水肿与液体积聚的重要标志。
临床公认吸入氧浓度60%为肺氧毒性的临界阈值,持续暴露会超出肺部内源性抗氧化防御能力,大量生成超氧阴离子、过氧化氢等活性氧。活性氧直接攻击I型、II型肺泡细胞膜脂质与蛋白质,引发脂质过氧化损伤;破坏表面活性物质合成,升高肺泡表面张力、降低肺顺应性,导致肺组织僵硬、通气阻力增加。
除生化损伤外,高浓度氧气会引发吸收性肺不张的力学改变:正常状态下氮气难以入血,可维持肺泡结构;当吸入氧浓度≥60%时,氧气替代氮气并快速被血液吸收,肺泡内压力缺失进而塌陷。氮气冲刷效应会减少有效气体交换面积,诱发高氧性急性肺损伤,病理特征与急性呼吸窘迫综合征相似,表现为非心源性肺水肿,远期可进展为肺纤维化。
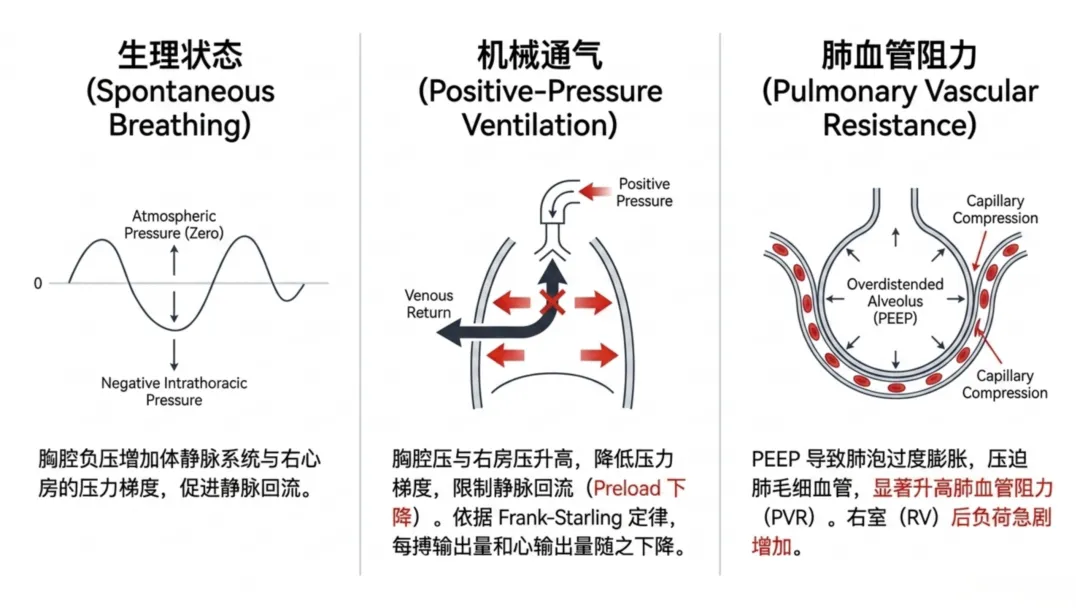
机械通气会改变胸腔内压力环境,直接调控心脏负荷状态。自主呼吸时,胸腔负压增大体循环静脉与右心房压力梯度,促进静脉回流;正压通气则升高胸腔内压与右心房压,减小压力梯度、限制静脉回流。跨壁压为心腔压力与胸腔周围压力的差值,是评估心肌壁张力的核心指标;胸腔内压升高可降低有效心脏负荷,且不受体循环动脉压影响。
机械通气会随呼吸周期造成心肌壁张力周期性改变,重塑心肌应变模式,激活参与结构重构的机械敏感信号通路。除短暂影响心输出量外,机械通气可从根本上改变心肌应激的力学调控机制,参与心脏长期结构适应性改变。
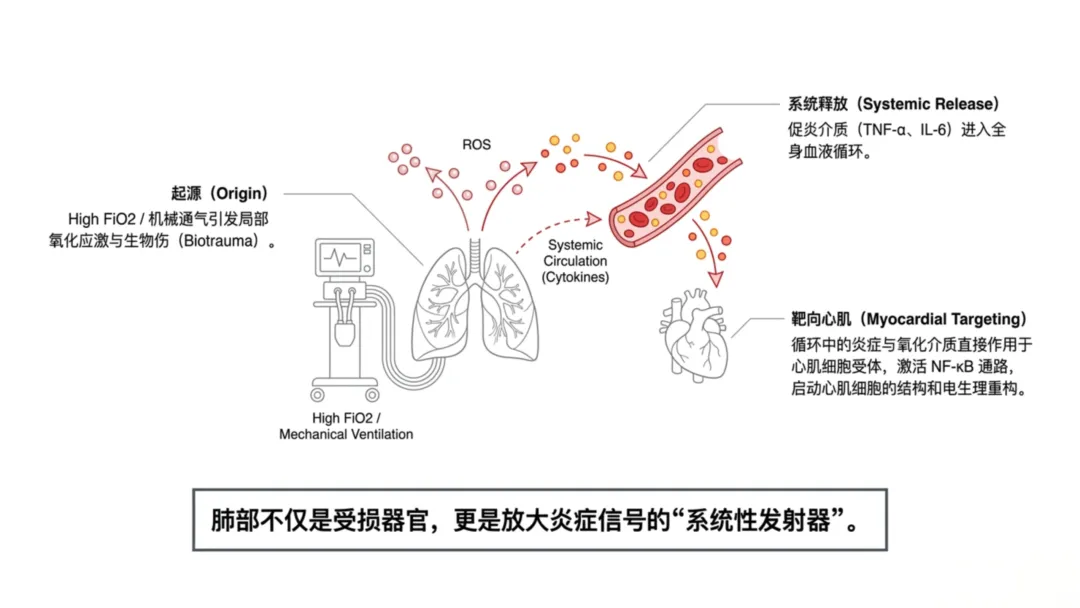
[图1 心肺-活性氧轴与高氧诱导心血管重构机制图]
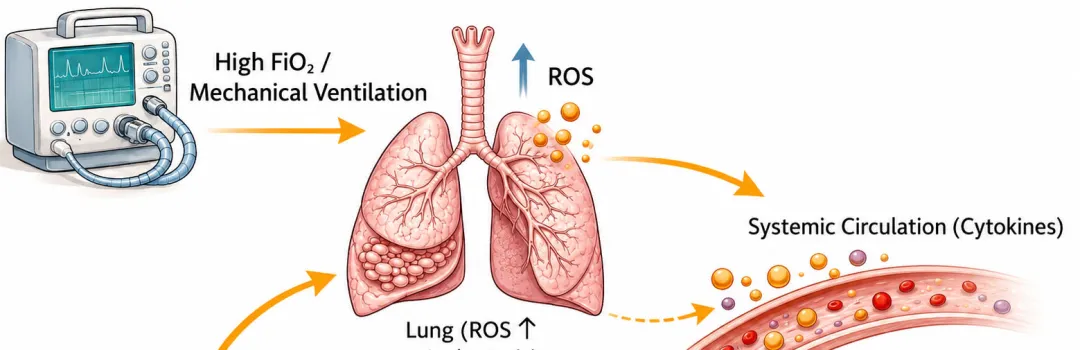
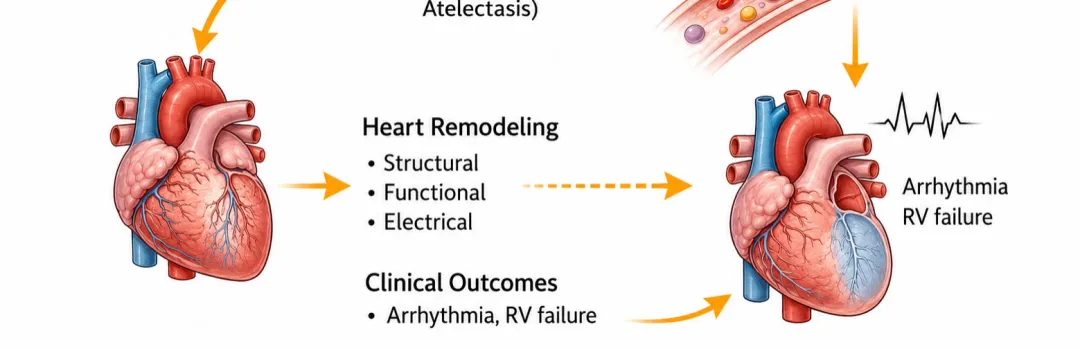 图1 心肺-活性氧轴与高氧诱导心血管重构机制:示机械通气下胸腔内压升高、肺血管阻力增加、活性氧生成、炎症信号激活及心肌结构/电生理重构的级联通路
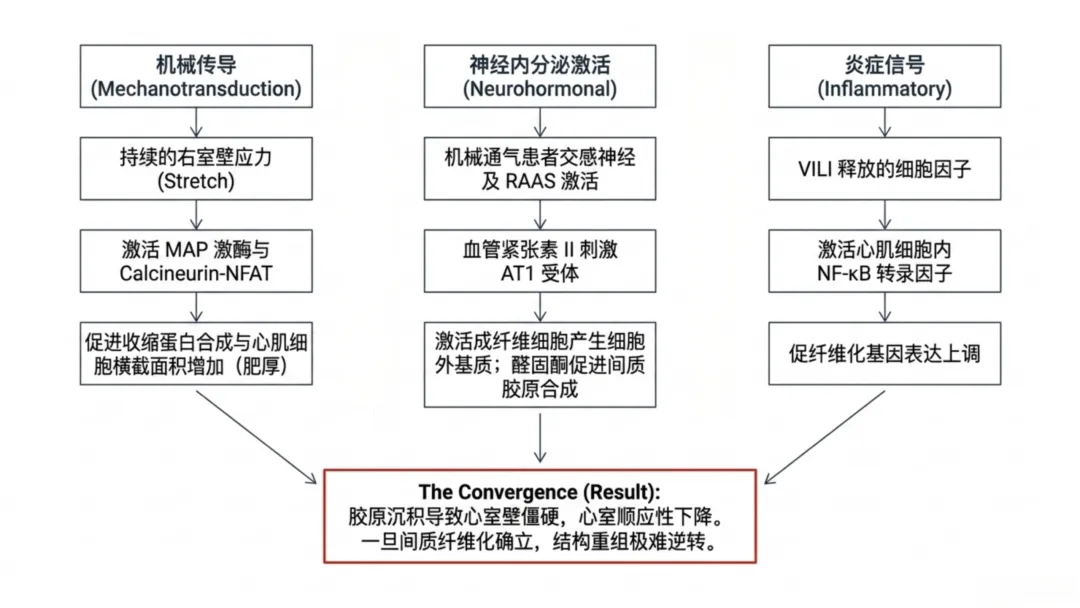
图1 心肺-活性氧轴与高氧诱导心血管重构机制:示机械通气下胸腔内压升高、肺血管阻力增加、活性氧生成、炎症信号激活及心肌结构/电生理重构的级联通路机械通气使心肌承受反复机械牵拉,心肌细胞通过固有机械敏感信号通路产生应答。牵拉敏感离子通道与细胞骨架可将壁张力变化转化为细胞内生化信号;心室壁负荷升高会激活丝裂原活化蛋白激酶、钙调磷酸酶-活化T细胞核因子、磷脂酰肌醇3-激酶-蛋白激酶B等信号通路,调控基因转录。动物实验证实,转录水平适应性改变会促进收缩蛋白合成与肌节扩增,增大心肌细胞横截面积。持续机械负荷会使适应性应答转变为持续性肥厚重构,将机械应激固化为心肌结构表型。
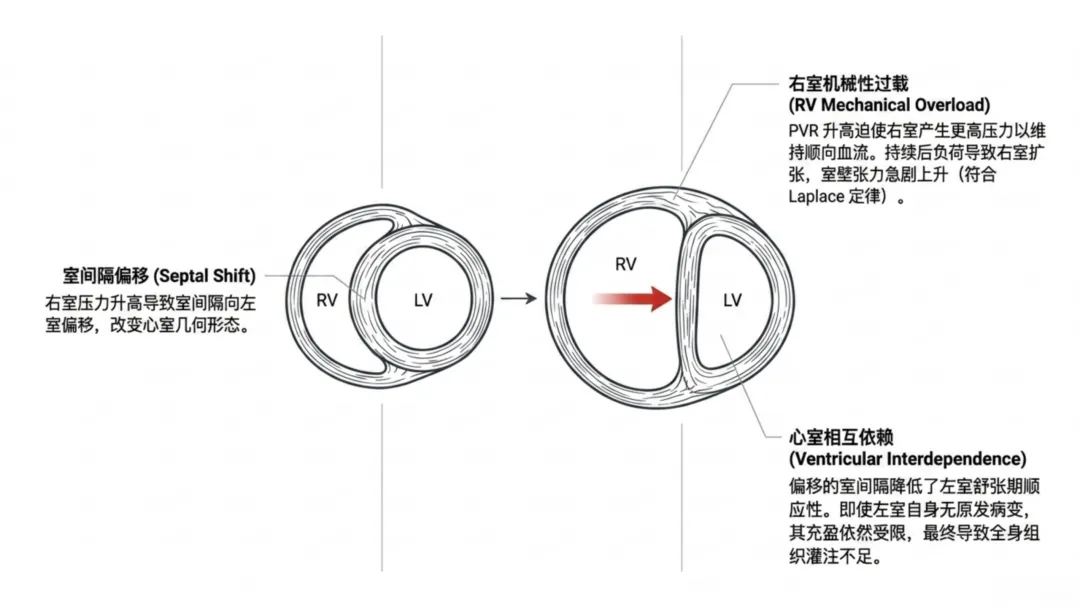
右心室对后负荷变化高度敏感,机械通气时呼气末正压升高、肺泡过度扩张会压迫肺毛细血管,升高肺血管阻力。肺血管阻力升高迫使右心室增高压力维持射血,依据拉普拉斯定律进一步增大室壁张力。初期右心室可通过增强收缩力、肥厚重构代偿以平衡壁张力;持续高后负荷会使代偿转为失代偿,引发心腔扩张、壁张力进一步升高,机械做功效率持续下降。
右心室重构会通过心室相互作用影响左心室功能:右心室压力升高、心腔扩张会使室间隔左移,改变心室几何形态,降低左心室舒张顺应性、重分布心肌壁张力。心内膜下组织受累尤为明显,持续机械牵拉会激活左心室肥厚与纤维化信号通路,即便无原发性左心室病变也会发生重构。因此,机械通气诱发的肺血管应激可通过力学耦合继发左心室重构。
神经体液激活是机械通气下心肌结构重构的另一重要途径。通气支持患者常伴随交感神经兴奋、肾素-血管紧张素-醛固酮系统激活。持续儿茶酚胺暴露会增加心肌细胞钙循环与代谢负荷,诱发心肌肥厚,长期刺激可启动凋亡信号通路。血管紧张素II通过激活1型受体促进肥厚基因表达,刺激成纤维细胞生成细胞外基质;醛固酮则促进心肌间质胶原合成,最终导致细胞外基质扩增、心肌僵硬度增加。胶原沉积会降低心室顺应性、损害舒张功能、升高充盈压,改变心肌壁张力分布,加重失代偿性重构,诱发功能与电生理不稳定。
呼吸机相关性肺损伤引发的炎症信号,是机械通气影响心肌结构的关键通路。肺泡过度牵拉或高氧暴露会促使促炎介质释放入血,将肺损伤效应扩散至全身循环。肿瘤坏死因子α、白细胞介素6等循环细胞因子可作用于心肌细胞与心脏成纤维细胞受体,激活核因子κB等细胞内信号通路,调控心脏组织基因表达,上调肥厚与细胞外基质合成相关基因,抑制正常收缩功能基因。该表达模式会促进成纤维细胞活化、心肌间质胶原沉积,加剧细胞外基质扩增;胶原累积会升高心肌僵硬度、破坏心肌细胞结构排列、削弱同步收缩功能,进一步降低心室顺应性、加重重构进展。
氧化应激会进一步加剧心肌结构重构。机械通气中常采用高氧维持动脉氧合,会在肺及全身组织大量生成活性氧。心肌细胞内氧化应激会破坏线粒体膜结构、损伤电子传递链功能,降低线粒体效率、减弱氧化磷酸化、减少三磷酸腺苷生成。
能量供应是维持心肌功能的基础,心肌收缩时肌动蛋白-肌球蛋白横桥循环、舒张时肌浆网钙重摄取均依赖三磷酸腺苷。三磷酸腺苷生成不足会导致舒张过程能量代谢障碍、舒张功能恶化;慢性能量应激还会激活代偿性信号通路,促进心肌结构重构与肥厚型适应性改变。氧化损伤由此将代谢应激与心脏结构改变紧密关联。
活性氧还可直接调控细胞外基质重构,通过氧化信号激活促纤维化转录因子,促进成纤维细胞向肌成纤维细胞分化,加速间质纤维化与心室壁僵硬。同时,氧化应激会影响冠脉微循环,降低一氧化氮生物利用度、损伤内皮功能;内皮功能障碍会损害冠脉微循环灌注,诱发亚临床微血管缺血,进一步激活炎症与纤维化信号,形成"氧化应激-纤维化-灌注受损-氧化应激加重"的恶性循环。
氧暴露时长决定机体应答为适应性还是失代偿性重构:短期机械通气仅造成前负荷、后负荷短暂改变,无长期结构损伤;而持续肺血管应激、炎症激活、神经体液信号及氧化损伤会形成重构前馈循环。随着肥厚进展与细胞外基质含量增加,心腔形态改变、室壁增厚或扩张、顺应性下降,舒张充盈功能受损;间质纤维化一旦形成难以逆转,属于组织结构重塑而非短暂细胞应答。因此,长期机械通气即便撤除呼吸支持,仍可能对心肌造成持续性结构损伤。
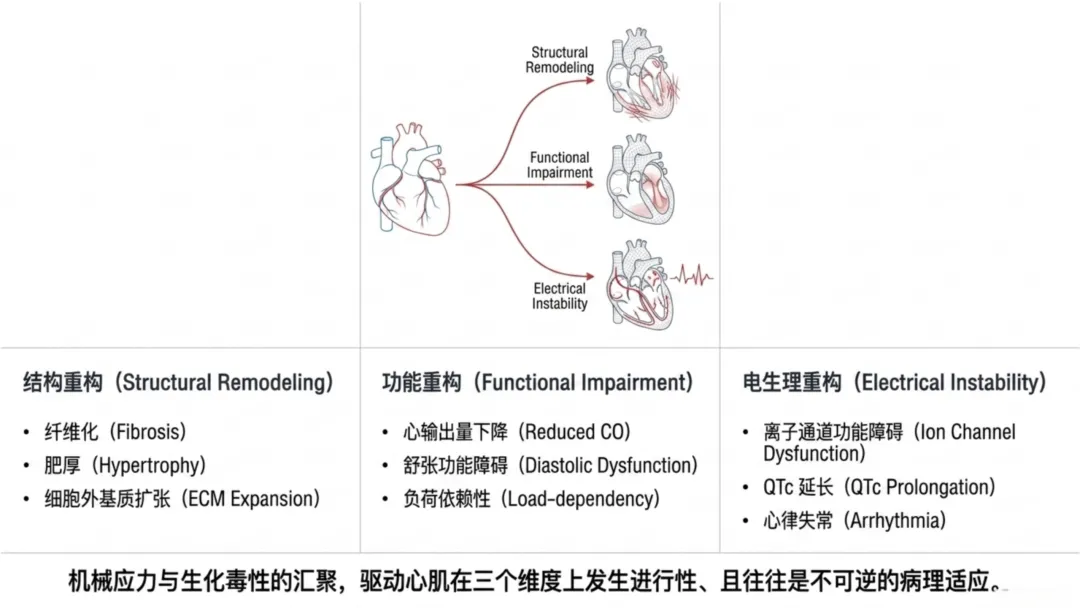
综上,机械通气通过力学、血管、神经体液、炎症、氧化多重机制调控心脏结构,改变负荷状态、基因表达、线粒体功能及细胞外基质组成,最终形成以僵硬度升高、心腔形态异常、机械效率下降为特征的进行性重构,为后续功能损伤与电生理不稳定奠定病理基础。
机械通气可从临床层面显著改变心功能,而心输出量是决定全身氧供的核心因素。即便氧合正常,心输出量下降仍会导致组织灌注不足、缺氧。危重症患者前负荷轻度降低或右心室后负荷升高,即可显著减弱全身灌注、加重休克、诱发器官损伤。机械通气会造成心脏负荷周期性变化,逐次改变心室充盈与射血功能,呼吸机参数可依据患者基础生理状态影响心功能表现。
静脉回流由体循环平均充盈压与右心房压力梯度决定,正压通气升高胸腔内压与右心房压,减小压力梯度、限制静脉回流。前负荷下降会通过弗兰克-斯塔林机制降低每搏量,进而减少心输出量。容量不足患者该效应尤为显著,基础充盈压低,胸腔内压小幅升高即可导致静脉回流大幅下降。脓毒症、脱水、失血及强效利尿患者,上调呼气末正压或平均气道压时易出现低血压、灌注受损。静脉回流减少会降低心输出量与全身氧供,若氧供低于代谢需求,即便肺部氧合正常,仍会发生组织缺氧与器官功能损伤,出现呼吸指标改善而全身灌注恶化的矛盾现象,提示需结合血流动力学综合评估呼吸机参数调整。
除降低前负荷外,机械通气可通过升高肺血管阻力增加右心室后负荷。肺血管阻力与肺容积、肺泡压力密切相关,肺泡过度扩张压迫肺毛细血管,通气不良区域通过缺氧性血管收缩进一步升高阻力。肺血管阻力升高迫使右心室增高压力维持射血,初期可通过增强收缩力代偿,持续高后负荷会降低每搏量、诱发心腔扩张,升高壁张力、增加氧耗、降低机械效率。右心室射血量决定左心室前负荷,右心室射血减少会继发左心室充盈不足、全身心输出量下降。该交互作用提示右心室功能在机械通气中的重要性,呼吸机参数可通过升高后负荷、限制前向血流诱发易感患者循环不稳定。
机械通气升高右心室壁张力还会促进心室重构、加重心室相互作用异常,进一步限制左心室充盈与全身灌注。机械通气过程中肺动脉压力升高、右心室负荷过重,与危重症患者血流动力学紊乱及不良临床预后密切相关。
舒张功能是机械通气状态下心功能的重要决定因素。胸腔内压升高、前负荷降低会减少心室充盈容积;肥厚、纤维化、炎症所致心室顺应性下降患者,充盈量小幅减少即可引发每搏量显著降低。钙调控异常会进一步损害舒张功能、升高舒张充盈压。此类人群中,机械通气可暴露或加重舒张功能障碍、耗尽充盈储备,即便收缩功能正常,仍会出现心输出量受限,在舒张功能高发且易漏诊人群中尤为突出。
超声心动图可评估机械通气患者心功能,但需结合负荷状态解读。射血分数具有负荷依赖性,后负荷降低时可表现为正常或偏高,即便每搏量已下降。因此,射血分数正常或高动力状态可与充盈受限、前向血流受损并存。舒张功能指标也会随胸腔内压、静脉回流改变,无法单纯反映心肌固有舒张异常。每搏量、左心室流出道速度时间积分、三尖瓣环收缩期位移及室间隔运动等指标,可更直接评估血流动力学状态。右心室扩张、室间隔扁平、前向血流减少,提示压力负荷过重与心室交互异常;血流呼吸变异度、充盈压升高可进一步反映机械通气对心功能的影响。
机械通气通过改变胸腔压力环境与右心室射血阻力,调控静脉回流、右心室后负荷、心室交互作用及最终心输出量。心输出量下降会损害器官灌注与氧供,加重休克病理状态及多器官功能损伤,即便肺部氧合有所改善。因此,机械通气的心血管效应是呼吸机管理的核心内容,对急性呼吸窘迫综合征、脓毒症、前负荷储备不足及右心室功能受损患者尤为重要。
心肌电活动依赖心肌细胞膜离子通道的有序激活与失活。心动周期由快速去极化启动,经精准时序复极化恢复膜电位稳态,为下一次兴奋做准备。该过程的完整性取决于离子通道密度及门控动力学特征。机械通气不会直接对心脏施加电刺激,但会改变离子通道的生理微环境;全身炎症、氧化应激、机械牵拉及体温波动,会逐步调控离子通道表达与传导功能,间接重塑心肌电生理特征。
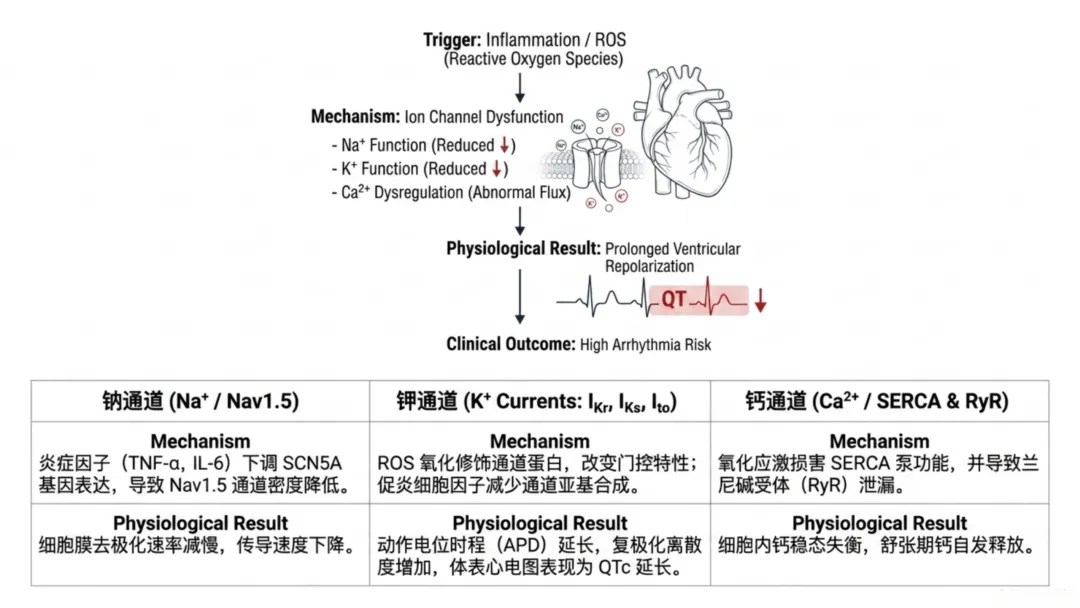
心室动作电位快速上升支由钠通道介导,主要由SCN5A基因编码的Nav1.5通道调控。钠电流大小决定去极化上升斜率,进而影响心肌组织传导速度。高效电传导依赖肌膜功能性钠通道密度及快速激活动力学特征。长期机械通气患者体内肿瘤坏死因子α、白细胞介素6等炎症介质水平升高,细胞因子与心肌受体结合后激活细胞内信号通路,调控离子通道基因转录。持续炎症信号会降低钠通道表达、抑制通道膜转运,减少功能性通道数量、降低峰值钠电流。
钠电流减弱会减慢细胞膜去极化速率、降低传导速度,导致心室心肌电传导空间异质性。电兴奋波前会遭遇部分复极化的心肌区域,形成折返环路,是持续性室性心律失常的核心机制。因此,机械通气过程中钠通道轻度下调,即可形成易诱发心律失常的传导病理基础。
动作电位复极化过程主要由快速延迟整流钾电流、缓慢延迟整流钾电流、内向整流钾电流、瞬时外向钾电流等外向钾电流调控。复极化时长决定不应期,协调心肌兴奋性同步恢复,均匀复极化可保障心室同步舒张、避免过早再次兴奋。炎症介质与活性氧可直接影响钾通道表达与传导功能:氧化修饰改变通道蛋白门控特性,细胞因子介导转录调控降低钾通道亚基合成,最终导致外向复极化电流密度下降。
钾电流减少会延长动作电位时程与不应期,临床心电图表现为QT间期延长。组织层面的影响更为关键:复极化过程并非均匀延长,而是出现区域恢复时间差异,不应期离散度产生电位梯度,诱发异常电兴奋传导。复极化延长还会在动作电位2、3期诱发早期后除极。本团队率先开展高氧诱导心脏电生理与结构重构研究,证实不同年龄、性别个体在高氧暴露下均会出现QTc间期显著延长及缓慢性心律失常。机械通气患者联用延长QT间期药物时,该效应会进一步放大。
[图2 高氧与炎症应激下心脏电重构及心律失常发生机制图]
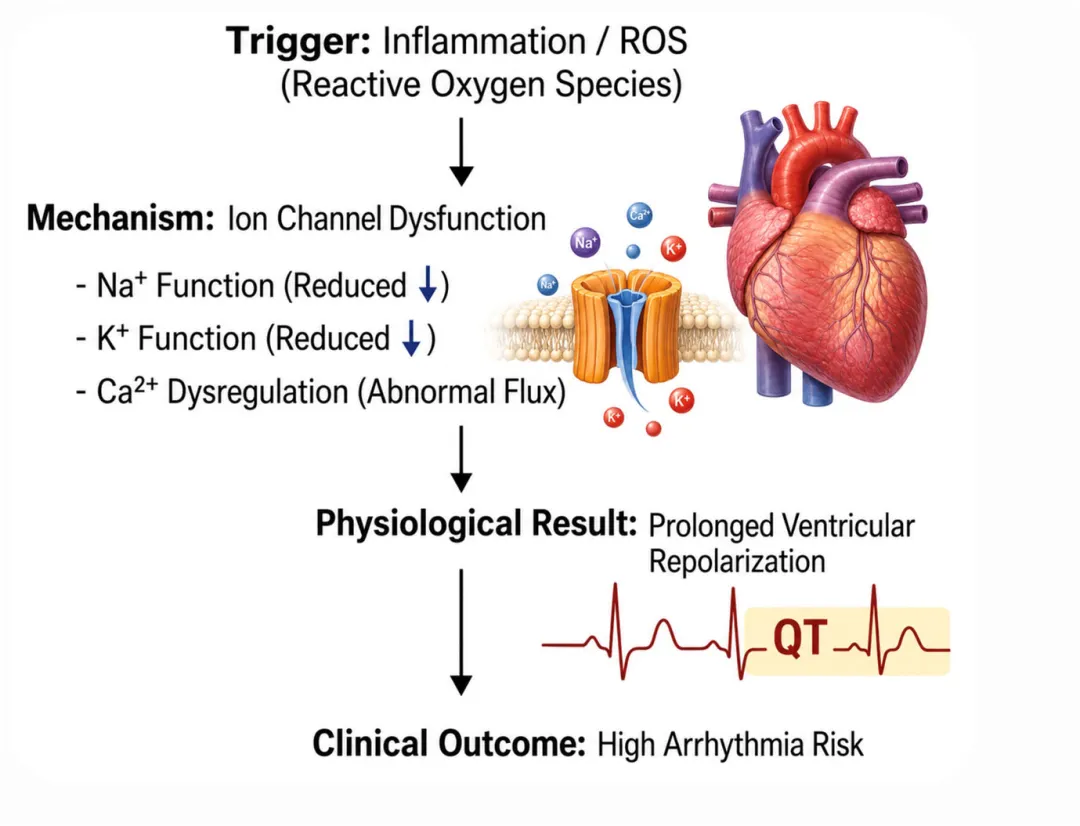 图2 高氧与炎症应激下心脏电重构及心律失常发生机制:示钠通道下调致传导减慢、钾通道功能异常致复极化离散、钙稳态失衡诱发延迟后除极,以及机械牵拉激活机械敏感通道的综合效应
图2 高氧与炎症应激下心脏电重构及心律失常发生机制:示钠通道下调致传导减慢、钾通道功能异常致复极化离散、钙稳态失衡诱发延迟后除极,以及机械牵拉激活机械敏感通道的综合效应心肌电兴奋稳态还依赖胞内钙循环的精准调控。动作电位期间钙内流触发肌浆网钙进一步释放,启动心肌收缩;舒张期钙经肌浆网钙三磷酸腺苷酶依赖泵体重摄取。氧化应激会损害肌浆网钙三磷酸腺苷酶功能、增加雷诺丁受体渗漏,导致钙重摄取障碍、钙自发性释放增加,破坏胞内钙稳态。
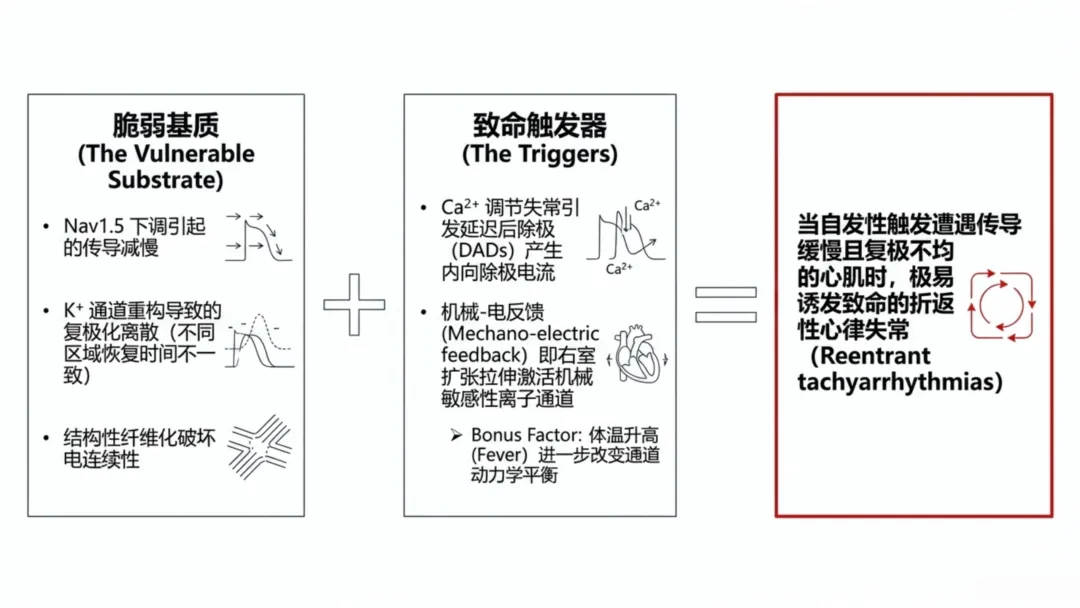
舒张期钙自发性释放会激活钠钙交换体,产生内向去极化电流,诱发延迟后除极。延迟后除极可作为电触发因素,诱发室性早搏。危重症患者交感神经兴奋会进一步升高胞内钙负荷,增加机械通气患者触发式去极化风险。当此类电触发因素存在于传导减慢、复极化离散的心肌组织中,易诱发持续性心律失常。
机械牵拉是电不稳定的另一重要机制。机械通气升高右心室后负荷、增大室壁张力,心肌细胞膜牵拉激活机械敏感离子通道,产生去极化电流、降低兴奋阈值。该机械应激可独立于炎症与氧化应激直接诱发电活动异常,即机电反馈效应,体现结构负荷与膜兴奋性的内在关联。
体温变化可进一步调控离子通道动力学特征,钠、钾通道的构象转变具有温度依赖性。体温升高会加快通道激活与失活速率,发热是机械通气危重症患者常见表现,可改变动作电位形态。由于离子通道表达及代谢应激存在区域差异,体温波动会增大复极化异质性,不仅加快心率,还可从分子层面改变去极化与复极化电流平衡。
心律失常发生需同时具备触发因素与易感基质。机械通气下触发因素包括钙稳态失衡所致延迟后除极、机械牵拉诱发去极化;易感基质包括钠通道功能异常所致传导减慢、钾通道重构所致复极化离散,纤维化还会进一步破坏电传导连续性。机械通气使机械牵拉、炎症信号、氧化损伤、代谢应激、体温调控共同作用于离子通道系统,叠加诱发电生理不稳定,而非单一因素独立致病。
机械通气自新冠疫情初期便成为核心治疗手段,长期临床随访数据证实,有创机械通气会加重、加速心肌重构。新冠心血管登记研究数据显示,住院期间合并心肌梗死的新冠患者,接受有创机械通气的比例显著更高;此类患者康复后心肺再入院风险也明显升高,且该关联独立于其他混杂因素。
拉丁美洲队列研究显示,合并心肌损伤的新冠患者接受机械通气后死亡率居高不下;除80岁以上高龄外,有创机械通气是此类患者院内死亡的最强独立危险因素,风险权重高于慢性肾病等基础疾病。超声随访研究发现,危重症新冠患者入院时无右心室损伤,气管插管机械通气后31%会新发右心室功能障碍,直接证实机械通气与新冠患者新发心脏损伤的关联。
其核心机制为:有创机械通气持续高呼气末正压会人为升高胸腔内压、压迫肺毛细血管、增加肺血管阻力,使薄壁结构的右心室负荷骤增、进行性功能衰退。随着通气时长增加,肺部高压持续作用会引发右心室进行性扩张、心室耦合解离,且该改变在插管初期并未出现。
机械通气联合新冠感染会产生协同效应,显著加重心肌重构与心脏损伤:呼吸机压力负荷造成右心室机械过载与牵拉应激,活性氧激活生物信号通路引发细胞损伤。临床需对新冠患者采取更保守的机械通气策略,严格控制生理血氧分压水平,减轻心脏损伤、降低死亡率。
动物实验明确阐明了高氧影响心肺生理的分子机制,新冠及危重症临床队列研究也证实了高氧暴露的死亡风险与远期重构危害。如表1所示,辅助供氧与高氧诱发的病理生理改变会影响患者远期预后,临床应将辅助供氧视为具有剂量窗口的药物性干预手段。
临床需加强监测,及时识别QT、QTc间期延长等供氧相关异常;将目标氧浓度下调至更保守标准,减少活性氧下游代谢通路激活。诊疗中需采用最低有效剂量滴定供氧,频繁评估病情、尽早撤机减氧;通过限制氧浓度与暴露时长,减轻氧化应激所致肺水肿与心肌重构。
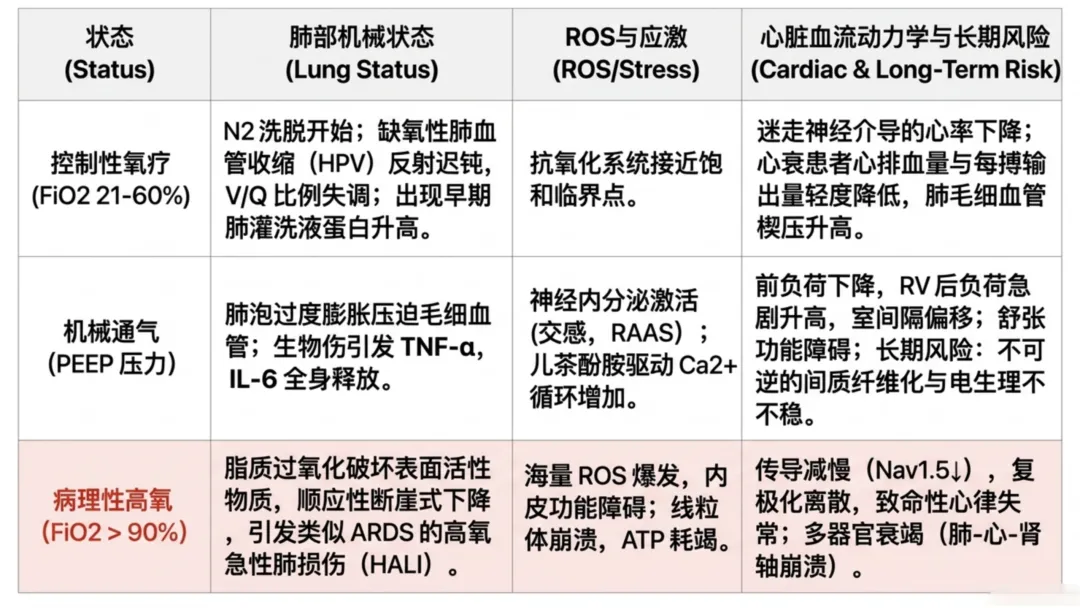
| 分类 | 驱动因素 | 肺部状态 | 活性氧/应激水平 | 心脏效应 | 远期风险 |
| 正常氧合(基线) | | | 氧化还原平衡、谷胱甘肽与超氧化物歧化酶抗氧化能力充足 | | |
| 可控性辅助供氧 | | 缺氧性肺血管收缩反射减弱→通气/血流比例失衡;肺泡灌洗液蛋白升高提示早期损伤;氮气减少诱发肺泡早期不稳定;50%~60%浓度持续7天可使肺组织易感氧毒性;炎症模型中肺重量升高提示肺水肿 | 低浓度吸氧时抗氧化系统充足,接近60%浓度时抗氧化能力逐渐耗竭 | 迷走神经兴奋减慢心率→心脏指数下降;每搏量降低;肺毛细血管楔压升高;心力衰竭患者易出现血流动力学负荷过重 | |
| 机械通气(压力支持) | | 氮气冲刷→吸收性肺不张;肺泡过度扩张→毛细血管受压;呼吸机相关性肺损伤引发生物创伤,释放肿瘤坏死因子α、白细胞介素6入血 | 机械牵拉应激;神经体液激活:交感神经兴奋、肾素-血管紧张素-醛固酮系统激活;儿茶酚胺升高钙循环负荷 | 静脉回流与前负荷降低;肺血管阻力升高→右心室后负荷增加;室间隔移位→左心室充盈减少;舒张功能障碍、心室僵硬致心输出量受限;射血分数可表现正常但每搏量降低,机械通气下射血分数参考价值有限;血管紧张素II促进肥厚基因表达;醛固酮促进胶原合成 | 右心室扩张、间质纤维化(纤维化形成后难以逆转);电生理不稳定;进行性结构重构 |
| 病理性高氧血症 | | 脂质过氧化→I/II型肺泡细胞破坏;表面活性物质功能异常→表面张力升高、肺顺应性下降;高氧性急性肺损伤→类急性呼吸窘迫综合征损伤,远期进展为肺纤维化;动物模型持续暴露72~96小时死亡率升高 | 大量活性氧生成;一氧化氮减少→内皮功能障碍;核因子κB激活→肌成纤维细胞分化;线粒体功能障碍→三磷酸腺苷生成减少;肌浆网钙三磷酸腺苷酶功能下降+雷诺丁受体渗漏→钙稳态失衡 | 心输出量与射血分数降低;心动过缓;QTc、JT间期延长;钠通道表达下降→传导减慢、折返性心律失常;钾通道功能异常→延迟后除极;室间隔移位、右心室衰竭;活性氧激活肌成纤维细胞、重构离子通道→复极化离散、钙稳态紊乱 | 间质纤维化、心律失常发生;纤维化形成后损伤难以逆转;急性肾损伤与心肺肾多器官功能障碍;新冠患者插管后31%新发右心室功能障碍;机械通气是80岁以下患者院内死亡首要独立危险因素 |
注:↑ 表示升高/增加;↓ 表示降低/减少;→ 表示导致/引发。
※ 左右滑动可查看完整表格内容
现有文献研究存在一定局限性:高氧诱导心肌结构与电生理重构(离子通道下调、炎症细胞因子激活)的机制研究多基于动物实验,难以将动物模型的氧损伤时间-剂量阈值直接推演至危重症人群,临床结局常受基础疾病混杂干扰。
机械通气与辅助供氧患者常同时接受多种药物治疗,难以单独区分高氧血症与药物对重症患者心律失常的贡献,尤其部分药物本身可延长心脏复极化过程。临床常用射血分数等负荷依赖性心功能指标,易漏诊高氧诱导的心肌功能损伤,需推广左心室流出道速度时间积分、三尖瓣环收缩期位移等非负荷依赖指标,精准评估正压通气下血流动力学状态。
基础研究证实高氧暴露可对心肌造成永久性结构印记,但仍缺乏长期临床随访研究,无法明确一过性高氧暴露对存活患者远期慢性心力衰竭发病风险的影响。
辅助供氧是现代医学不可或缺的治疗手段,但过度应用会引发显著心血管不良结局。本文综述证实,高氧血症通过氧化应激、炎症反应及机械通气相关力学紊乱,成为心肺功能障碍的核心驱动因素。过量活性氧会破坏线粒体功能、扰乱钙稳态、激活促纤维化与肥厚信号通路,共同推动心肌结构重构。
同时,胸腔内压力改变、肺血管阻力升高会造成血流动力学应激,尤以右心室受累显著,最终损害心输出量与全身组织灌注。上述结构、功能改变叠加电生理重构,为心律失常发生提供病理基础。
重要的是,高氧损伤存在剂量与时间依赖性,临床亟需转变诊疗理念,推行保守化、滴定式精准氧疗,在保障组织氧供的同时最大程度减轻高氧损伤。未来研究需聚焦最佳氧合阈值界定、人群易感差异分析,研发靶向干预措施以减轻氧化与炎症损伤,改善危重症患者心血管远期预后。
| 缩略词 | 英文全称 | 中文全称 |
| AKI | | |
| ARDS | Acute respiratory distress syndrome | |
| ATP | | |
| COPD | Chronic obstructive pulmonary disease | |
| COVID-19 | | |
| ECM | | |
| EF | | |
| FiO2 | Fraction of inspired oxygen | |
| HALI | Hyperoxic acute lung injury | |
| HFNC | | |
| HPV | Hypoxic pulmonary vasoconstriction | |
| ICU | | |
| LV | | |
| LVOT VTI | Left ventricular outflow tract velocity–time integral | |
| MV | | |
| NIV | | |
| PaO2 | Partial pressure of arterial oxygen | |
| PEEP | Positive end-expiratory pressure | |
| PVR | Pulmonary vascular resistance | |
| RAAS | Renin–angiotensin–aldosterone system | |
| ROS | | |
| RV | | |
| SaO2 | Arterial oxygen saturation | |
| SERCA | Sarco/endoplasmic reticulum calcium ATPase | |
| TAPSE | Tricuspid annular plane systolic excursion | |
| TNF-α | Tumor necrosis factor alpha | |
| VILI | Ventilator-induced lung injury | |
| V/Q | | |
※ 左右滑动可查看完整表格内容
通过网盘分享的文件:辅助氧疗对心血管的生理影响.pdf
链接: https://pan.baidu.com/s/1zQVWVDUMiNwG51MD8BG32Q?pwd=e7cn 提取码: e7cn
本文内容译编自专业学术文章,仅供重症医学专业人员学习参考,不作为临床诊疗依据。内容均参考原始文献整理并经人工审核校对,如有歧义请以原文为准,原文版权归原作者及出版方所有,本次中文编译整合仅用于学术交流,严禁商用及未经授权转载。